IgA血管炎
IgA血管炎は、以前はSchonlein Henoch紫斑病として記載されていた血管炎で、IgA沈着を特徴とする小血管の全身性血管炎です。1802年にWilliam Heberdenによって、関節炎、紫斑病、胃腸および腎臓の病変を患う4歳の男児についての最初の記述がなされました。1837年にJohann Schonleinは紫斑と関節痛の関係を認識し、1874年にEduard Henochは胃腸管と腎臓の関与を追加しました。以後、この疾患はSchonlein Henoch紫斑病としてよく知られていました。2012年CHCC(Chapel Hill Consensus Conference)において、IgA血管炎という呼称が採択され、Schonlein Henoch紫斑病という呼称は廃止されました。この疾患は小児において最も一般的な全身性血管炎であり、通常は合併症がなく、自然に治まる経過をたどります。成人ではIgA血管炎になる頻度は低くなりますが、腎臓や胃腸の症状が現れる場合には、経過はより複雑になります。典型的な症状は、触知可能な紫斑、関節痛または関節炎、腸炎および糸球体腎炎です。IgA血管炎の長期予後は腎臓の重症度に影響されます。
<注記> ドイツの総説を主に参考にしていますので、日本の実情とは異なる可能性があります。
疫学
IgA血管炎は小児(4-7歳がピーク)における最も一般的な全身性血管炎であり、年間発生率は10万人あたり3〜27.2人であると推定されています。成人の場合、年間発生率は10万人あたり0.8〜2.2人と推定されています。平均発症年齢は50歳です。女性患者よりも男性患者(1:1.5)の方がわずかに多くなっています。
誘因
IgA血管炎は、感染症、悪性疾患、自己免疫疾患、または薬剤の使用に関連して発症する可能性があります。 小児では季節的に秋から冬に症例が増加し、ほとんどの場合、上気道、下気道、胃腸管に既存の感染症が存在しています。対照的に、IgA血管炎と感染症との関連は成人ではあまり観察されません。一般的な病原体には、β-連鎖球菌(30〜50%の症例で検出されます)、黄色ブドウ球菌、ヘリコバクター・ピロリ、マイコプラズマ肺炎、水痘、アデノウイルス、パルボウイルスB19、B 型肝炎ウイルス、サイトメガロウイルス、EBウイルスなどがあります。COVID-19感染に続発するIgA血管炎の症例(4)の文献では23例)も報告されています。IgA血管炎はCOVID19ワクチン(4)の文献では20例、私も1例経験しています)を含むワクチン接種後にも観察されています。薬剤関連IgA血管炎としては、抗生物質 (β-ラクタム系抗生物質、フルオロキノロン、マクロライド) および TNFα阻害剤との関連を示されています。悪性疾患(特に固形悪性腫瘍)との関連性も報告されています。悪性腫瘍関連IgA血管炎患者は高齢であり、壊死性紫斑病、血尿、肺胞出血、全身症状がより頻繁にみられます。成人でIgA血管炎が長期にわたって難治性の経過をたどっている場合に悪性腫瘍の合併に注意する必要があります。さらに、IgA血管炎と炎症性腸疾患、脊椎関節炎、家族性地中海熱との関連性も報告されています。
病態生理
IgA血管炎の正確な病態生理学はまだ解明されていません。IgA血管炎は、IgA1が血管内で免疫複合体を形成し、その免疫複合体が血管内に沈着する小血管の白血球破壊性血管炎です。血液中を循環するIgA1はグリコシル化(特にガラクトシル化)が減少しているため、ガラクトース欠損IgA1 (Gd-IgA1) と呼ばれます。その特異な生化学的特性により、Gd-IgA1は組織内に沈着しやすい傾向があるのですが、さらにGd-IgA1と抗Gd-IgA1-IgG自己抗体、補体などが結合し、免疫複合体を形成し組織内沈着が進みます。Gd-IgA1は、IgA血管炎患者の腎臓と皮膚で検出される可能性があります。ゲノムワイド関連研究 (GWAS) によって明らかにされたIgA血管炎との関連がある遺伝的素因は、HLA クラス II 遺伝子 (HLA-DQ および HLA-DRB1 を含む) 、インターロイキン(IL-8、IL-18 を含む)などの免疫メディエーター、内皮機能の調節因子 (eNOS)、補体系、腸のバリア機能に関与する遺伝子などです。
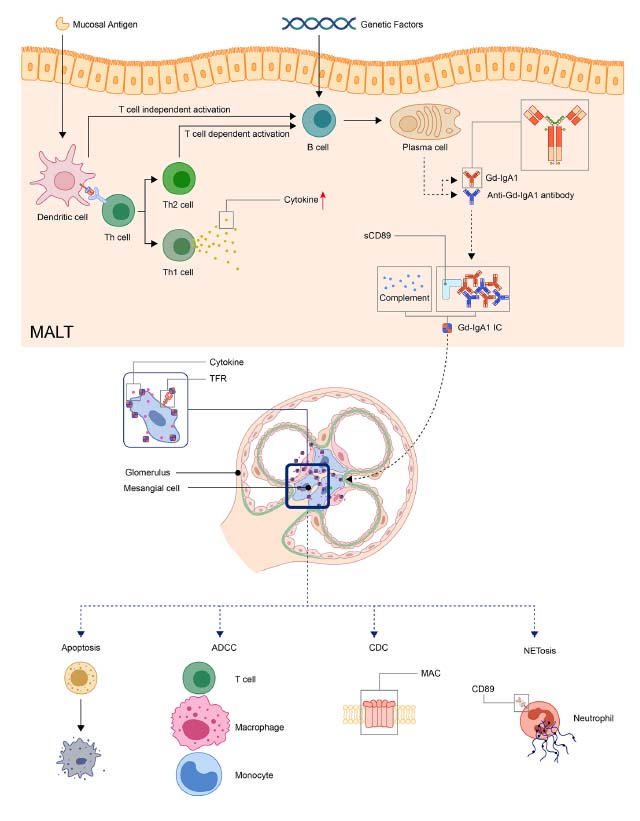
IgA 血管炎の病因
粘膜抗原は、T細胞依存性または非依存性にMALTのB細胞を活性化できます。遺伝的要因により、活性化された B細胞は形質細胞になり、Gd-IgA1を産生します。 Gd-IgA1および抗Gd-IgA1自己抗体は、補体、sCD89とともに免疫複合体を形成します。次に、免疫複合体が臓器に沈着し、炎症反応を活性化します。 腎臓では、免疫複合体が TfR を通じてメサンギウム細胞を活性化し、腎細胞のアポトーシスと炎症細胞の補充を引き起こす可能性があります。 (ADCC、抗体依存性細胞毒性; CDC、補体依存性細胞毒性; Gd-IgA1、ガラクトース欠損 IgA1; MAC、膜攻撃複合体; MALT、粘膜関連リンパ組織; NET、好中球細胞外トラップ; TfR、トランスフェリン受容体)
Pathogenesis of IgA Vasculitis:An Up-To-Date Review
Front. Immunol. 12:771619. doi: 10.3389/fimmu.2021.771619
症状
IgA血管炎の古典的な3主徴には、血小板減少症または凝固障害のない触知可能な紫斑、関節痛/関節炎、および腹痛が含まれます。典型的な3主徴が存在する場合、特に小児では、IgA血管炎の診断は臨床的に行うことができます。
皮膚(日本での発現率 100%)
皮膚症状は触ると少し隆起した状態がわかる紫斑(palpable purpura)が特徴的です。触知可能な紫斑は、病初期には症例の約75%で見られ、疾患の経過全体では患者の100%に見られます。小児では下肢の領域で発生し、成人では場合によっては上肢や体幹にも発生します。皮膚血管炎は徐々に進行し、約2-3週間続いた後、軽快します。成人の約30%が壊死性または出血性紫斑病を経験します。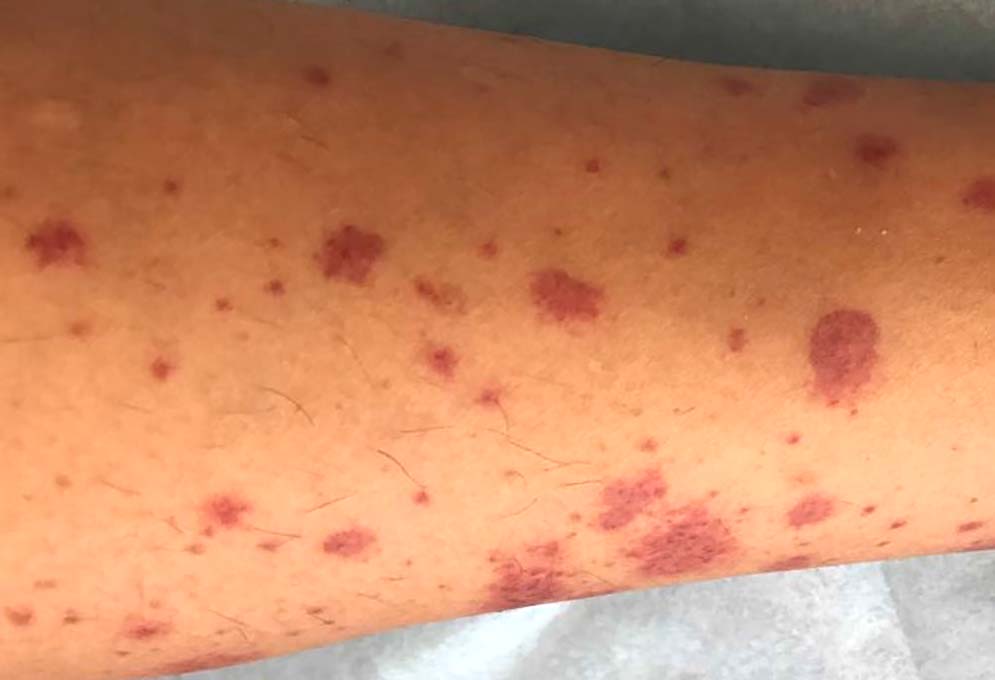
関節(日本での発現率 60〜75%)
関節痛および関節炎は、小児ならびに成人IgA血管炎患者の約84%で報告されています。通常、下肢の大きな2つか3つの関節が侵され、急性に発症します。関節破壊は見られません。筋肉痛についても記載されています。
消化管(日本での発現率 50〜65%)
病気が進行すると(皮膚症状が出た後しばらくして)、半数以上の患者が疝痛のような腹痛を経験し、吐き気や嘔吐を伴うこともあります。患者の50%で便中に潜血が検出されることがあります。まれに、胃腸出血、虚血、さらには壊死、腸重積、穿孔を伴う重篤なケースが発生します。腸重積は特に小児によく見られます。消化器症状は紫斑の出現以前に出現することが稀ではなく(14〜36%)、本症の診断がなされないまま不要な外科手術が施行されることもあるので注意を要します。
腎臓(日本での発現率 20〜55%)
IgA血管炎に関連する腎症状は小児では、20〜50%で発症し、成人では76%と高頻度に認められます。腎症状は、紫斑や関節症状から少し遅れて(1か月以内)出現することがあるので要注意です。尿検査では、血尿、赤血球円柱および軽度のタンパク尿(<1g/24時間または1000mg/gクレアチニン:以後クレアチニンの記載を省きます)がみられます。小児における腎病変は軽度であり、通常は特別な治療を行わなくても完全に治癒しますが、成人では、病初期から30%の症例で腎機能障害を来たしており、再発率は20%、末期腎不全のリスクは11%と報告されています。そのため、腎生検は、診断を確定し、腎障害を評価するために非常に重要です。予後不良因子としては、1g/24時間を超えるタンパク尿、糸球体濾過量(GFR)低下、高血圧、腎生検における間質性の線維化と糸球体の硬化などが挙げられます。
IgA血管炎の腎臓症状と、IgA腎症には組織学的類似性があります。IgA腎症およびIgA血管炎を含む家族性クラスターも報告されています。IgA腎症は、糸球体内のIgA沈着およびメサンギウム増殖変化によっても特徴付けられますが、全身症状は見られません。ただし、IgA血管炎の場合の腎炎は急性腎炎/急速進行性糸球体腎炎/ネフローゼ症候群として現れますが、IgA腎症は通常徐々に進行し、無症候性血尿が特徴的です。腎症状合併のIgA血管炎患者はIgA腎症患者よりも若く、通常、腎機能がより急速に悪化します。この2疾患が異なる疾患であるか、または一連の疾患の異なる発現であるかについては、依然として議論の余地があります。
その他の症状
肺胞出血の有病率は0.8〜5%で、肺胞出血が合併した場合の死亡率は28%と報告されています。まれに心筋炎、不整脈、弁膜炎が報告されています。その他、精巣炎、精巣捻転、ぶどう膜炎、角膜炎、強膜炎、脳血管炎、末梢神経障害などの合併が報告されています。
検査
臨床検査
紫斑を認めますが、血小板数は正常で、凝固検査でも異常はありません。便潜血陽性、貧血は胃腸からの出血を示唆する所見です。赤沈値亢進やCRP高値などの炎症所見を認めることが多いですが、正常範囲内であることもよくあります。症例の50-70%で、血清中のIgA値の上昇が見られ、これは腎臓病変との関連があります。研究室レベルではガラクトース欠損IgA1 (Gd-IgA1) の測定が試みられています。疾患特異的な自己抗体はありません。PR3-ANCA、MPO-ANCA、抗核抗体は通常陰性です。
腎病変を合併した場合、尿検査にて、血尿、赤血球円柱およびタンパク尿を認めます。重症腎病変の場合、特に急性腎不全または急速進行性糸球体腎炎が存在する場合は、クレアチニン値の上昇/糸球体濾過量(GFR)低下を認めます。さらに、腎病変合併IgA血管炎の約15%に補体(C3、C4、CH50)の低下を認めます。
画像検査
著しい腹部症状を認める場合は画像検査(超音波検査、CT、MRI、胃ならびに大腸の内視鏡検査など)が必要となります。CTでは、腸壁の肥厚、拡張した腸ループ、壁内または粘膜下血腫などが認められます。胃ならびに大腸内視鏡検査では、炎症、出血、粘膜浮腫(下図)などを検出できます。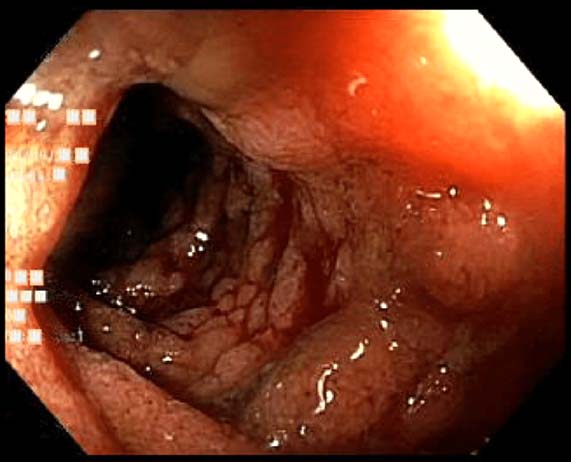
生検
典型的な臨床症状を示す小児では、診断を組織学的に確認する必要はありません。症状が非定型の場合、または、中程度から重度の腎病変を伴う場合は、生検を試みる必要があります。皮膚生検では、非特異的白血球破砕性血管炎の所見(下図)が見られ、直接免疫蛍光法によりIgA沈着物が示されますが、常に検出可能なIgA沈着物が伴うわけではありません。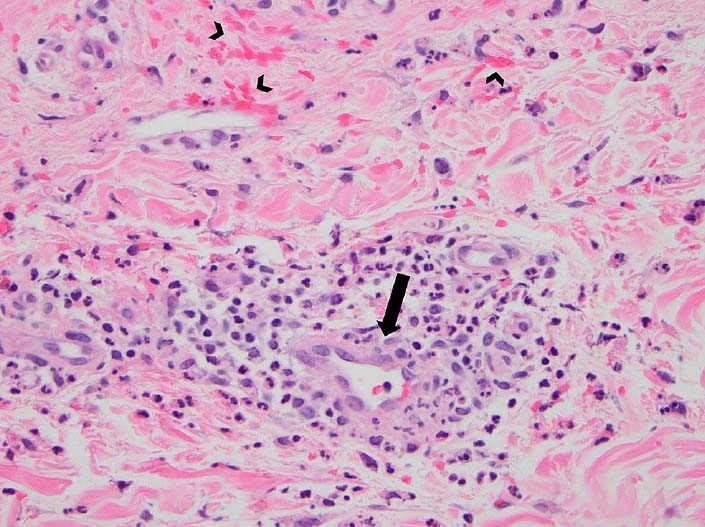
腎機能障害(糸球体濾過量低下)または1g/日以上の持続性タンパク尿がある場合、腎生検が必要となります。 組織病理学では、メサンギウム細胞の増殖と特徴的なメサンギウム領域へのIgA 沈着を認めます。活動性が上昇すると、糸球体毛細血管内の炎症細胞の増加や半月体形成を伴う糸球体壊死が検出される場合があります。
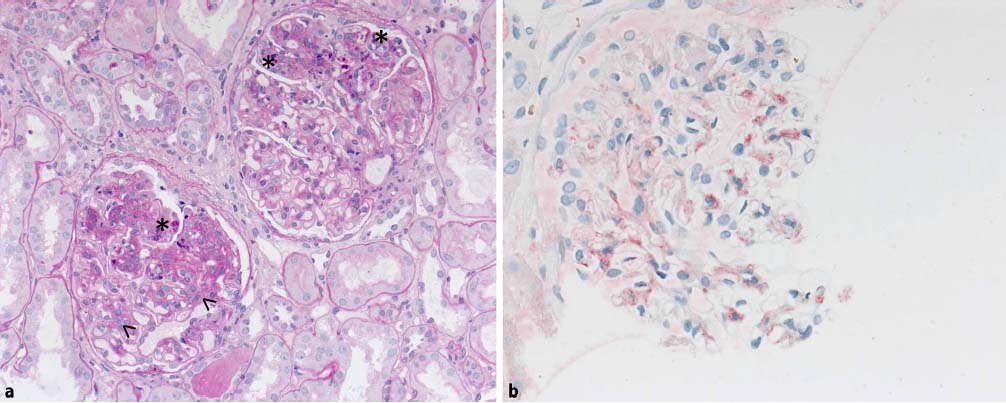
a 典型的な糸球体変化を伴う腎生検のPAS染色標本: 矢印: メサンギウム基質の増殖と細胞の増殖、アスタリスク: 毛細血管の細胞増殖。
b 免疫組織化学的 IgA 染色により、メサンギウム領域では明確な陽性所見(赤)が見られます。
診断と鑑別診断
IgA血管炎の診断は、特徴的な臨床症状に基づいて行われます。鑑別診断では、他の小血管の血管炎も区別する必要があります。IgA血管炎を分類するために、さまざまな分類基準が提案されています。1990年の ACR(American College of Rheumatology:米国リウマチ学会)分類基準では、基準のうち2つ以上が検出された場合、IgA血管炎は、約87%の感度と約87%の特異度で他の血管炎と区別できます。EULAR ( European League against Rheumatism:欧州リウマチ連盟)、PRINTO (Pediatric Rheumatology International Trials Organisation:小児リウマチ国際試験機構)、および PRES (Pediatric Rheumatology European Society:欧州小児リウマチ学会) による共同会議にて、小児におけるIgA血管炎の分類基準が2010 年に発表されました。EULAR/PRINTO/PRES基準では、小児において感度100%、成人でも感度99.2%、特異度86% であることが証明されています。2012年CHCC(Chapel Hill Consensus Conference)の定義では、IgA血管炎を小血管内に主にIgA1の沈着を伴う血管炎として定義しています。
| アメリカリウマチ学会の分類基準(1990年) |
|
1. palpable purpura:少し隆起した触知可能な紫斑で、出血性の皮疹、血小板減少を伴わない |
| 上記のうち、2項目を認めればIgA血管炎と分類できる。 |
| EULAR/PRINTO/PRESの分類基準(2010年) | |
| (血小板減少性紫斑病によらない)下肢優位の触知可能な紫斑もしくは点状出血 | 必須 |
|
1. 腹痛:急性に発症する腹部全体の疝痛(腸重積・消化管出血があっても良い) |
いずれか1つ以上 |
| 上記のうち、2項目を認めればIgA血管炎と分類できる。 |
| CHCC分類(2012年) |
| 小血管レベル(主に毛細血管、細静脈あるいは細動脈)の血管炎の中の免疫複合体性血管炎(Immune complex vasculitis)に分類され、血管壁にIgAのサブクラスであるIgA1優位の免疫沈着がみられる血管炎です。典型例では皮膚、腸管に病変があり、関節炎を引き起こします。病理組織学的にIgA腎症と区別のつかない糸球体腎炎を合併することもあります。 |
管理/治療
特に小児におけるIgA血管炎の管理は、自然軽快が多いことを考慮すると、対症療法が中心となります。治療アルゴリズムは臓器症状と病気の重症度に基づいています。治療選択肢にはグルココルチコイドや免疫抑制剤が含まれますが、エビデンスに関してはさまざまで、ほとんどが不十分な状況です。そのため、腎臓および胃腸の症状の治療は、ANCA関連血管炎およびIgA腎症に対する推奨事項に基づいて行われます。
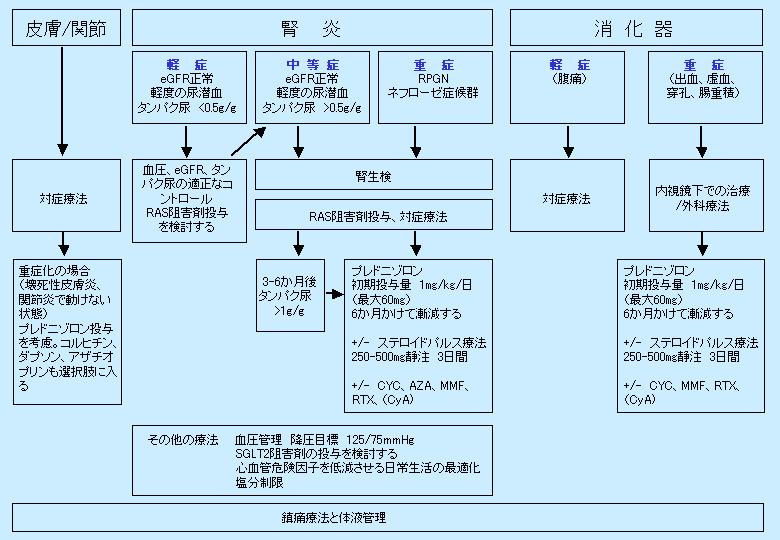
臓器症状と疾患の重症度を考慮したIgA血管炎血管炎の管理
1)の文献より引用改変RAS レニン・アンジオテンシン系、eGFR 推定糸球体濾過率、RPGN 急速進行性糸球体腎炎、CYC シクロホスファミド、AZA アザチオプリン、MMF ミコフェノール酸モフェチル、RTX リツキシマブ、CyA シクロスポリン A、SGLT ナトリウム-グルコース共輸送体
対症療法
対症療法には水分管理と適切な鎮痛療法が含まれます。鎮痛剤として、特に腎臓または胃腸症状の場合、非ステロイド性抗炎症薬 (NSAID) の使用は避けるべきです。アセトアミノフェンは使用可能です。ほとんどの症例は外来通院で治療可能です。重症の場合、特に重度の腎臓または胃腸障害、神経症状、静脈内補液の必要性、または重度の痛みの場合、入院が必要となる場合があります。
皮膚症状および筋骨格系症状の治療
ほとんどの場合、鎮痛剤(アセトアミノフェン)による治療で十分です。皮膚科医との連携も重要です。小児および成人におけるIgA血管炎の皮膚症状は通常約2〜4週間以内に回復します。ただし、壊死性皮膚炎や痛みのため動けない程の関節炎などの重篤な症状の場合は、グルココルチコイド療法を考慮する必要があります。専門家の意見によれば、用量はIgA血管炎の他の重篤な症状と同様に1〜2mg/kg (体重) (最大 60mg/日) とされています。通常、追加の免疫抑制剤の使用は必要ありません。現在治験進行中の治療法としては、コルヒチン、ダプソン、アザチオプリンが挙げられます。孤立した皮膚症状がある場合は、コルヒチン(開始用量0.5mgを1日2回、必要に応じて0.5mgを1日3回に増量)を使用できます。現在進行中の対照研究 (COLCHIVAS、NCT04008316) では、12か月の観察期間でコルヒチンによる治療の効果が調査されています。ダプソン(用量 1〜2mg/kg 体重または 50〜150 mg/日)使用前には、グルコース-6-リン酸欠乏症をチェックする必要があります。現在進行中の対照臨床試験 (ARAMIS、NCT02939573) では、IgA血管炎およびその他の皮膚血管炎に対するダプソン、コルヒチン、またはアザチオプリンによる治療法が研究されています。
胃腸症状の治療
軽度の場合は、腹痛に対する鎮痛療法と水分管理などの対症療法で十分です。出血、虚血、穿孔、または腸重積を伴う重篤な症例では、内視鏡下での治療および外科的治療が必要となる場合があり、消化器内科医、または外科医との連携が重要です。重症例では、グルココルチコイドまたは免疫抑制療法の使用も考慮されるべきです。
IgA血管炎関連腎炎の治療
IgA血管炎 に関連した腎臓症状の治療は、腎臓内科医との連携のもとに実施されるべきです。最初に、タンパク尿、糸球体濾過率 (eGFR)、および血圧に基づいて、疾患の重症度を評価します。腎臓の関与が最初は軽度であっても急速に進行する可能性があるから要注意です。腎臓の機能が正常で、軽度の尿異常(軽度の微小血尿、タンパク尿 <0.5g/g)の場合には腎生検は通常不要ですが、より重症度の高い場合(タンパク尿 (>0.5g/g) または、急速進行性糸球体腎炎)には(少なくとも成人では)腎生検を実施する必要があります。
IgA血管炎関連腎炎の対症療法
血管炎を伴わないIgA腎症と同様に、IgA血管炎関連腎炎の治療の重要な柱は、対症療法です。上記の一般的な対症療法に加えて、慢性腎臓病変の場合は、目標血圧値が 125/75mmHg 未満となるように血圧を最適化することに特に注意を払う必要があります。主に腎保護作用のあるRAS(レニン・アンジオテンシン系)阻害剤、すなわち、ACE (アンジオテンシン変換酵素) 阻害剤またはARB(アンギオテンシンII受容体拮抗薬)を使用し、最大許容用量まで徐々に増量します。慢性腎臓病では心血管のリスクが増加するため、心血管の危険因子とライフスタイルの評価と最適化(禁煙と体重管理など)も重要です。LDLコレステロールの管理も大事です。腎障害の程度に応じて、eGFR 30 〜 60の場合、LDLコレステロール目標値として、70mg/dl未満、さらにはeGFR <30の場合、55 mg/dl未満が適用されます。
軽度の腎炎(タンパク尿 <0.5g/g、軽度の尿潜血、eGFR正常)の場合は、血圧、eGFR、タンパク尿、LDLコレステロールの適正なコントロールをはかりながら、経過観察します。この段階で、ACE阻害剤やARBの使用も考慮に入れます。タンパク尿が0.5g/日を超える場合は、ACE阻害剤やARBの投与が必須です。eGFR低下や重大なタンパク尿を伴う場合は、SGLT-2阻害剤も投与する必要があります。最近の研究では、SGLT-2阻害剤はIgA腎症(血管炎なし)を含む慢性腎臓病患者の腎臓生存率も大幅に改善することが示されています。
中等症以上のIgA血管炎関連腎炎の導入療法
腎生検後、重症度が判明したら、重症度に即して寛解導入療法を開始します。IgA血管炎関連腎炎の寛解導入療法に関する質の高いランダム化対照臨床研究はほとんどないため、他の血管炎関連腎炎と同様の治療法に準じて治療します。以下の推奨は血管炎関連腎炎のガイドラインの推奨事項に、専門家の意見を加え補足したものです。
腎炎が中等度(eGFR正常、軽度の尿潜血、タンパク尿 >0.5g/g)の場合、最初の3〜6 か月間は対症療法で様子を見ます (ACE阻害剤やARBの使用、血圧管理、SGLT2阻害剤使用)。対症療法を行っても、タンパク尿が1g/gを超えて持続する場合は、グルココルチコイドの経口投与 (開始用量プレドニゾロン 1mg/kg、6か月かけて漸減) を検討します。
腎炎が重症で、急速進行性糸球体腎炎(腎機能障害が急速に進行しており、腎生検で糸球体壊死の割合が高く【半月体形成 >25%】になった状態)の場合、ANCA関連血管炎と同様に、非常に強力な免疫抑制導入療法を直ちに開始することが重要です。ステロイドパルス点滴療法の後に、シクロホスファミド(CYC)静注と組み合わせた経口グルココルチコイド(開始用量プレドニゾロン 1mg/kg、6か月かけて漸減)を考慮します。
一方、ネフローゼ症候群(浮腫、低アルブミン血症、タンパク尿 >3.5g/g)の臨床像があり、eGFRが正常範囲内に維持されている場合は、まずプレドニゾロン単独による経口療法を試みます。4〜8週間経っても反応がない場合は、シクロホスファミドの静注投与を考慮します。
上記の治療法に禁忌がある場合、または長期の高用量グルココルチコイド療法の副作用のリスクが高い場合は、リツキシマブ (RTX)、アザチオプリン、またはミコフェノール酸モフェチル (MMF) を使用することができます。例外的な場合には、シクロスポリン A (CyA) の使用が検討されます。シクロスポリン A (CyA) は、腎毒性が知られているため、腎臓障害の場合には慎重に使用する必要があります。
IgA血管炎関連腎炎維持療法
IgA血管炎の維持療法の重要性は不明です。ANCA関連血管炎と同様に、最初に重度の臓器疾患または生命を脅かす疾患の場合、アザチオプリンによる維持療法を12〜24か月間実施できます。
免疫抑制剤のIgA血管炎に対する効果
アザチオプリン(AZA)
IgA血管炎 を有する成人におけるAZA療法に関するデータはありませんが、小児患者を対象とした小規模な非対照研究のデータでは、腎炎の臨床経過が AZA によって改善されることを示してされています。さらに、皮膚の白血球破砕性血管炎にも反応が見られました。 皮膚症状に対する対症療法が効果がない場合、AZAはダプソンまたはコルヒチンの代替品として使用できます。
ミコフェノール・モフェチル(MMF)
いくつかの研究では、ANCA関連血管炎、または IgA腎症患者の寛解誘導におけるMMFの有効性が示されています。小児のグルココルチコイド抵抗性腎炎(ANCA関連血管炎、IgA腎症、IgA血管炎関連腎炎)における寛解の誘導と再発の予防を目的として、 MMFの投与を試みた2つの症例研究で有効性を証明された。IgA血管炎関連腎炎の成人患者におけるグルココルチコイド単独療法と比較したMMFの効果に関する後ろ向き研究結果は、全体的な寛解率は同じだが、MMFの方が反応が早く、副作用が少ないことを示していた。
シクロスポリンA(CyA)
CsAは、重度の腎障害のある24人の小児患者を対象とした対照研究で、メチルプレドニゾロンパルス後にプレドニゾロンを12か月間投与した単独療法群とCsA併用群とを比較して評価されました。 CsAは、プレドニゾロン単独療法よりも迅速な寛解導入と低い再発率を示しました。 成人患者については、症例報告が数件しかありません。
シクロホスファミド(CYC)
IgA血管炎関連腎炎の小児を対象とした唯一のランダム化研究では、対症療法と比べ、CYCの腎転帰に対するプラスの効果は示されませんでした。腎臓および胃腸に障害のある成人において、CYCは高用量のグルココルチコイドと比較した無作為化試験が行われたが、6か月後と12か月後の寛解率に有意差が見られませんでした。 ただし、これらの試験は症例数が少ないため、データの解釈には注意が必要です。
リツキシマブ(RTX)
RTX療法は、ANCA関連血管炎の寛解を誘導し維持するために使用されます。日本でも、ANCA関連血管炎に対し、RTXは保険適応となっております。難治性IgA血管炎を有する成人患者22名を対象とした研究では、6か月後の寛解率が91%であり、腎症状に対する良好な反応が示されました。
その他の治療法
5)の文献によると、濃縮ヒト血液凝固第XIII 因子製剤、免疫グロブリン大量静注療法、血漿交換、扁桃摘出は、残念ながら、最近目立った報告がないと記載されています。
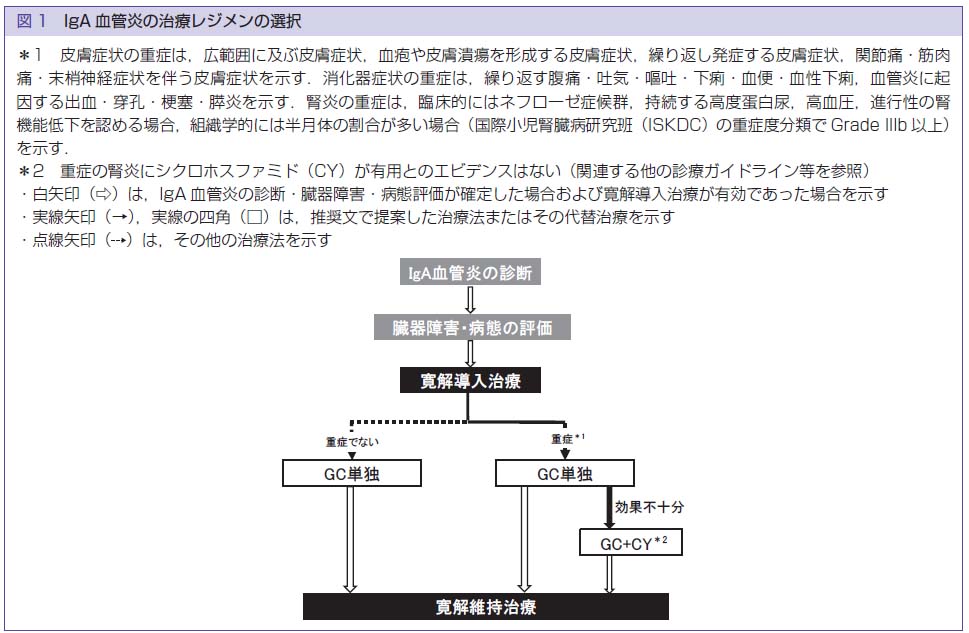
皮膚血管炎・血管障害診療ガイドライン2023によるIgA血管炎の治療レジメン
文献5)より
予後
小児のIgA血管炎は通常、合併症を伴わず、94%で完全に治癒します。成人の場合、IgA血管炎を有する患者の生存率は5 年後 73%、10年後 62%、20年後 45%と報告されています。特に、腎臓に病変がある場合、予後は著しく悪く、成人IgA血管炎250例のコホート研究は、32%が4カ月以内に腎機能不全を呈し,平均14.8年で持続性の腎機能異常が32%に見出され、そのうち11%が末期の腎不全、27%が中等度ないし重症腎不全に至ったと報告されています。成人患者の20〜40%で再発が報告されており、再発では腎外症状がより頻繁に発生します(胃腸16%、紫斑41%、関節痛11%)。
まとめ
・IgA血管炎 は、IgA免疫複合体形成を伴う小血管の全身性血管炎です。
・皮膚、関節、消化管、腎臓が影響を受ける可能性があり、その他の非常にまれな症状(肺胞出血、心筋炎、ぶどう膜炎、角膜炎、強膜炎、脳血管炎、末梢神経障害、睾丸炎)も発生することがあります。
・典型的な症状は、触知可能な紫斑、関節痛または関節炎、腸炎、糸球体腎炎です。
・IgA血管炎 は通常、自己制限的(特に小児では)であり、症状に応じた治療のみが必要です。 ただし、臓器または生命を脅かす合併症は、免疫抑制治療で積極的に治療する必要があります。
・IgA血管炎 の長期予後は腎臓の症状に影響されます。
文献
1)Immunglobulin-A-Vaskulitis
ZRheumatol https://doi.org/10.1007/s00393-023-01355-0 Angenommen:22.Marz 2023
2)Update Immunglobulin-A Vaskulitis
Z Rheumatol 2022 ・ 81:305-312, https://doi.org/10.1007/s00393-022-01162-z Angenommen: 30. Dezember 2021
3)Henoch-schonlein purpura following exposure to SARS-CoV2 vaccine or infection: a systematic review and a case report
Internal and Emergency Medicine 2023 https://doi.org/10.1007/s11739-023-03366-w
4) Pathogenesis of IgA Vasculitis:An Up-To-Date Review
Front. Immunol. 12:771619. doi: 10.3389/fimmu.2021.771619
5) 皮膚血管炎・血管障害診療ガイドライン2023―IgA 血管炎,クリオグロブリン血症性血管炎,結節性多発動脈炎,リベド様血管症の治療の手引き 2023―
日皮会誌:133(9),2079-2134,2023